- Algorithms & Theory
- Climate & Sustainability
- Conferences & Events
- Data Management
- Data Mining & Modeling
- Distributed Systems & Parallel Computing
- Economics & Electronic Commerce
- Education Innovation
- General Science
- Generative AI
- Global
- Hardware & Architecture
- Health & Bioscience
- Human-Computer Interaction and Visualization
- Machine Intelligence
- Machine Perception
- Machine Translation
- Mobile Systems
- Natural Language Processing
- Networking
- Open Source Models & Datasets
- Photography
- Product
- Programs
- Quantum
- RAI-HCT Highlights
- Responsible AI
- Robotics
- Security, Privacy and Abuse Prevention
- Software Systems & Engineering
- Sound & Accoustics
- Speech Processing
- Year in Review
-
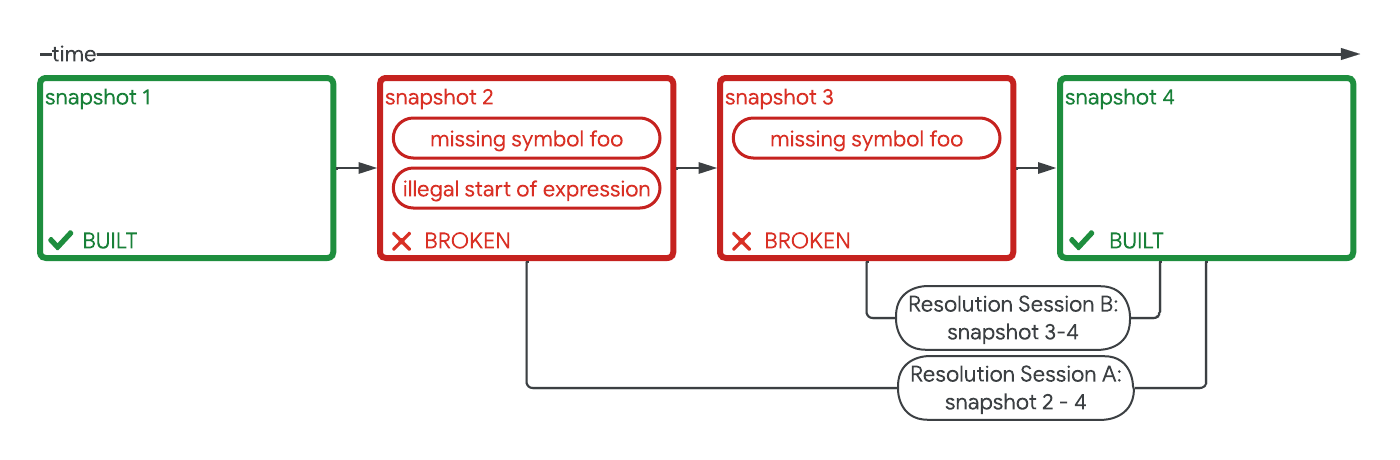
April 23, 2024
Safely repairing broken builds with ML- Machine Intelligence ·
- Software Systems & Engineering
-
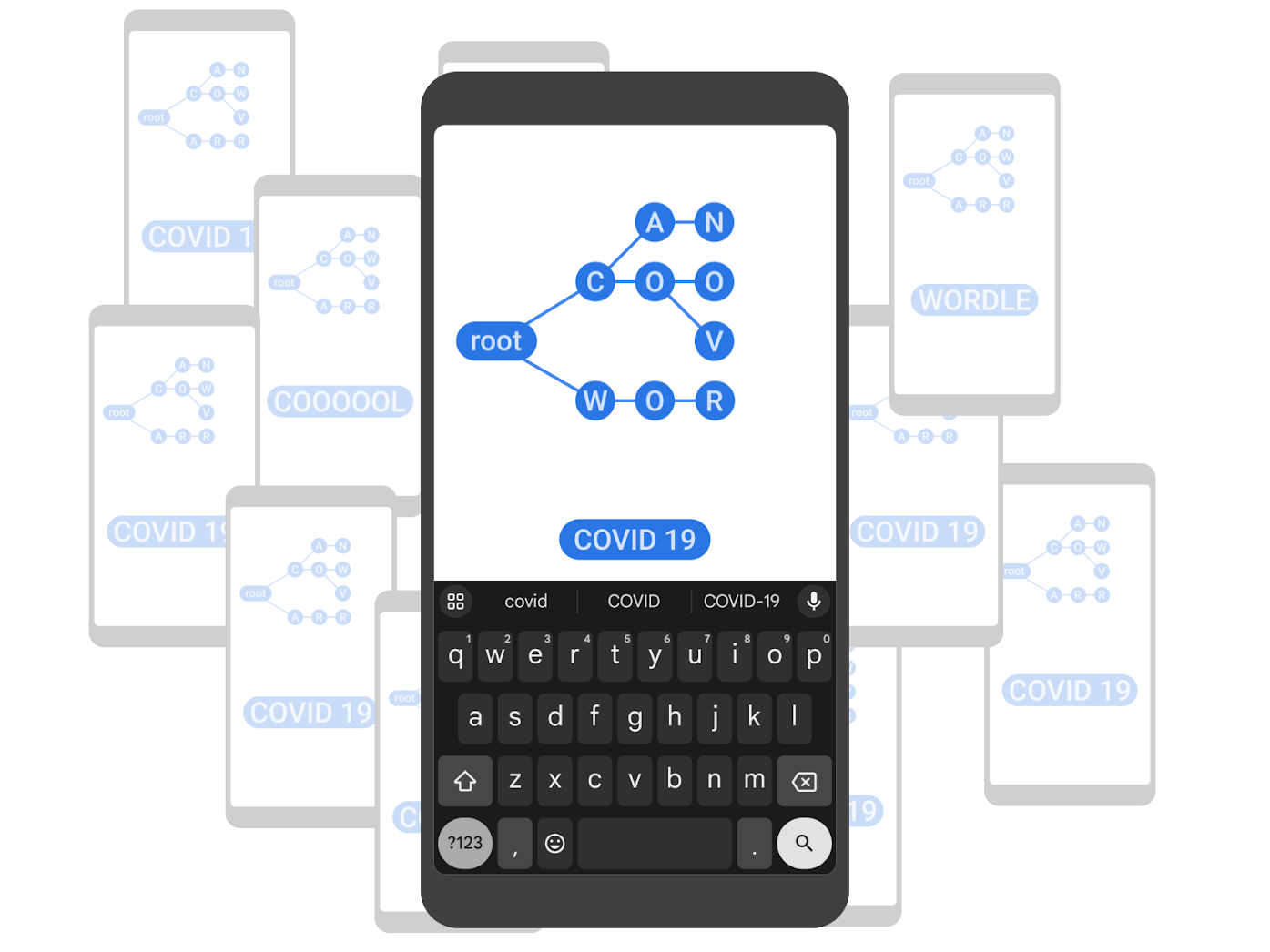
April 19, 2024
Improving Gboard language models via private federated analytics- Algorithms & Theory ·
- Distributed Systems & Parallel Computing ·
- Product ·
- Security, Privacy and Abuse Prevention
-
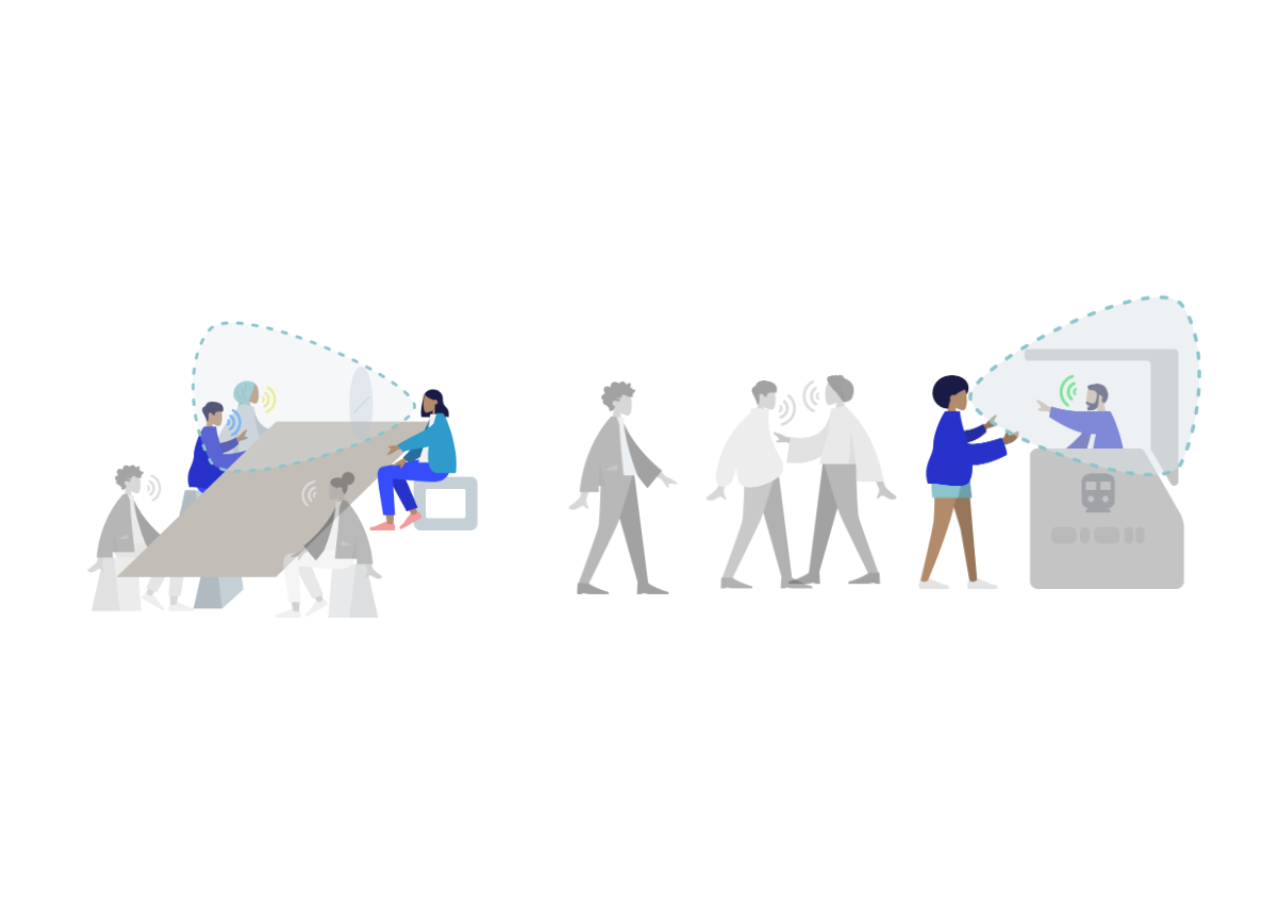
April 17, 2024
Robust speech recognition in AR through infinite virtual rooms with acoustic modeling- Human-Computer Interaction and Visualization ·
- Machine Perception ·
- Speech Processing
-

April 16, 2024
Solving the minimum cut problem for undirected graphs- Algorithms & Theory
-
April 12, 2024
Contrastive neural audio separation- Machine Intelligence ·
- Sound & Accoustics
-
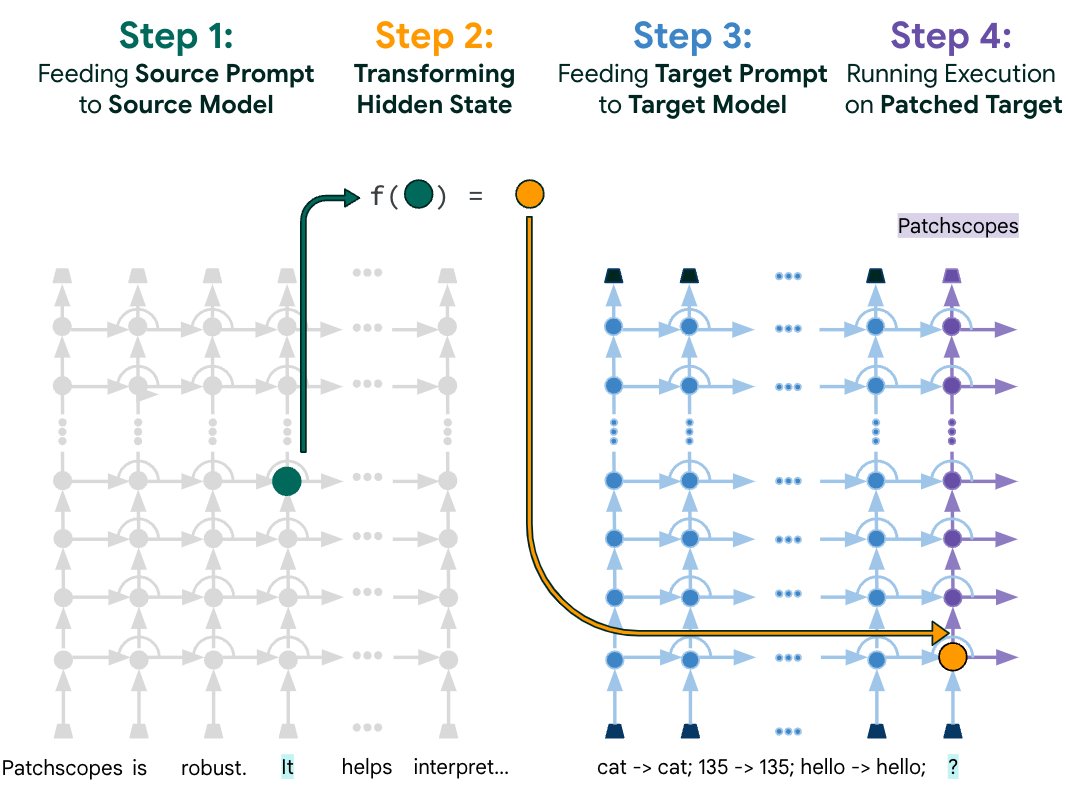
April 11, 2024
Patchscopes: A unifying framework for inspecting hidden representations of language models- Machine Intelligence ·
- Natural Language Processing ·
- Responsible AI
-
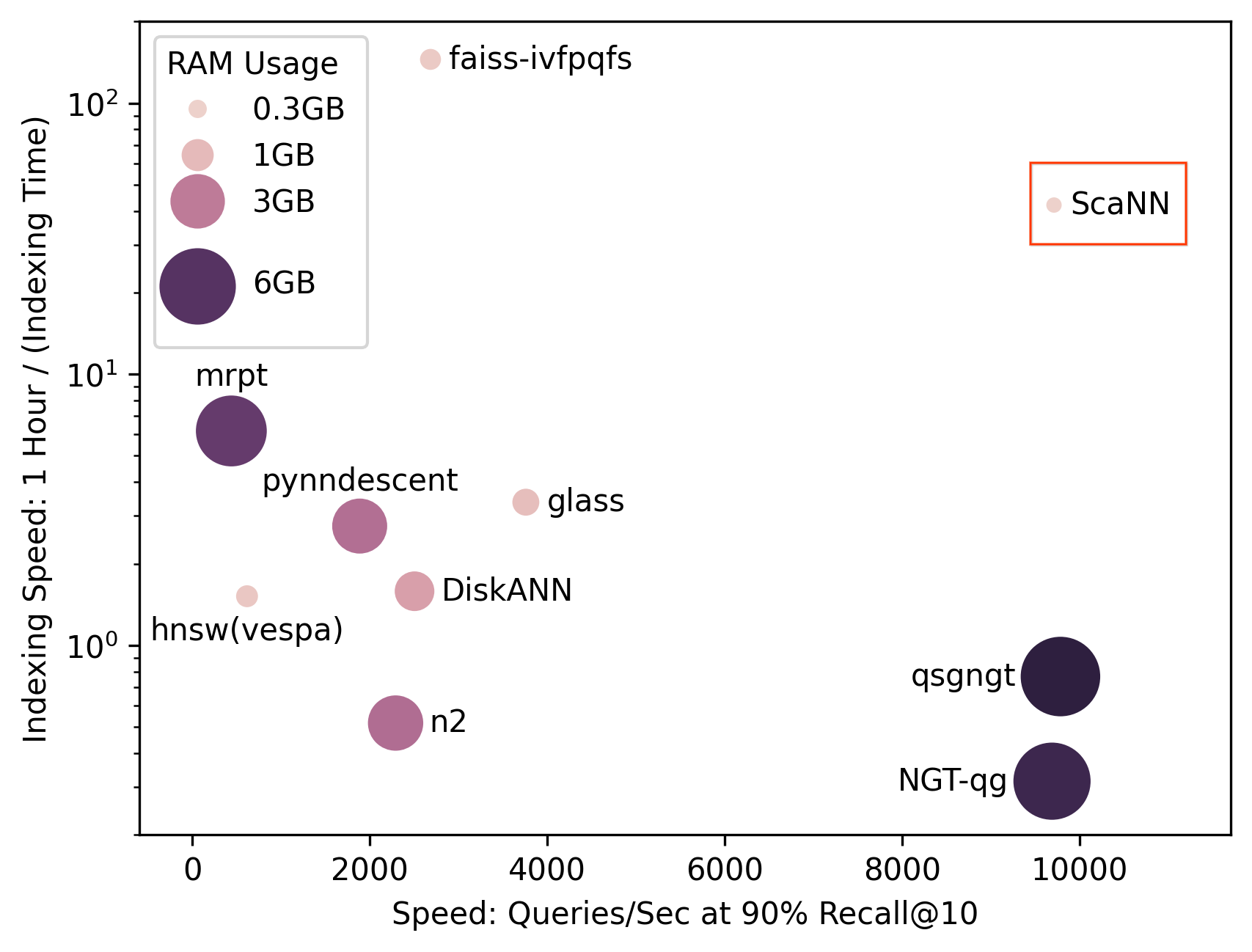
April 10, 2024
SOAR: New algorithms for even faster vector search with ScaNN- Algorithms & Theory ·
- Conferences & Events ·
- Data Mining & Modeling
-
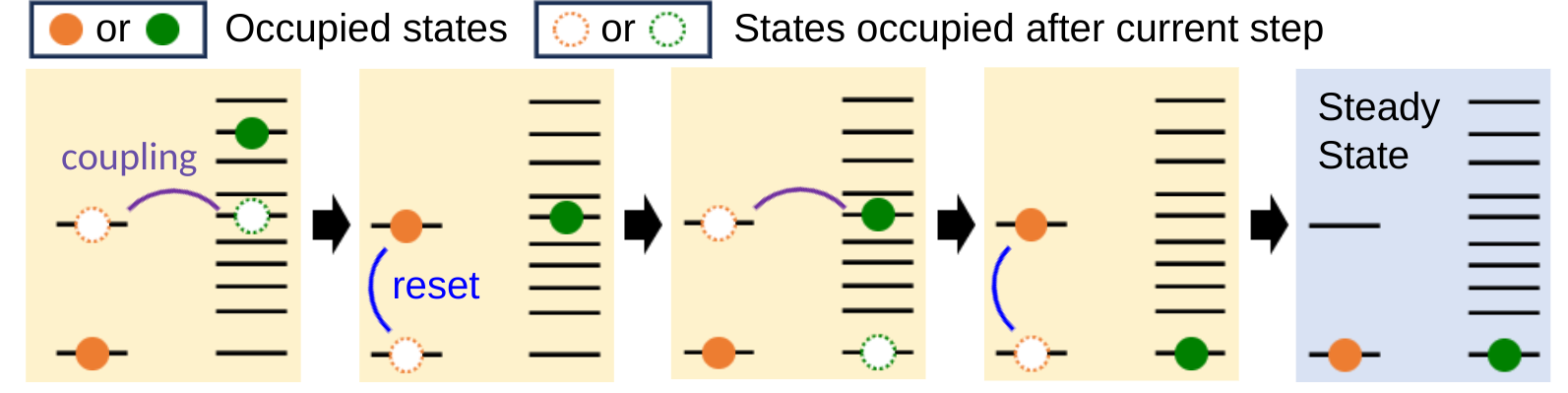
April 9, 2024
Preparing and stabilizing quantum states through engineered dissipation- Quantum
-
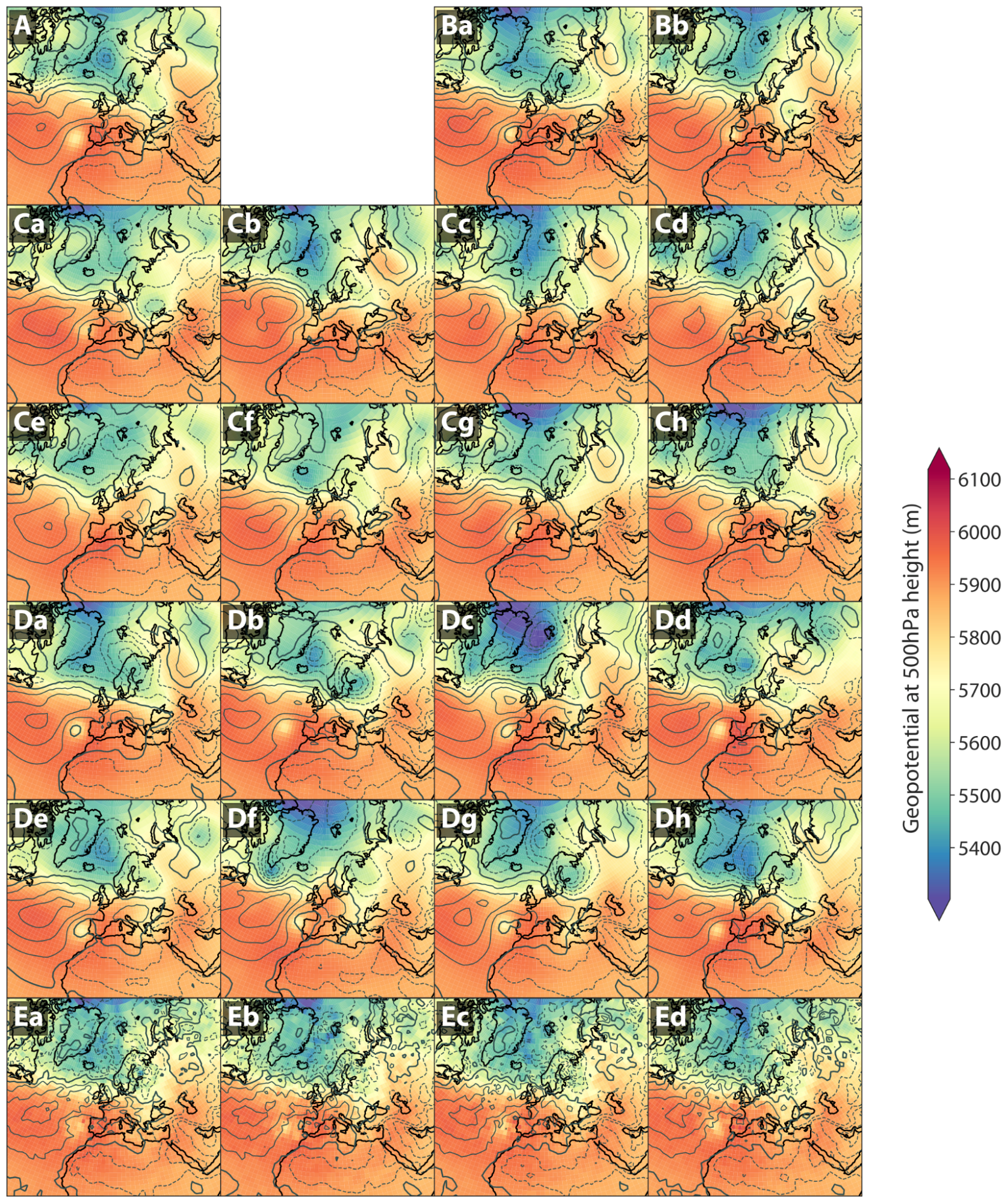
March 29, 2024
Generative AI to quantify uncertainty in weather forecasting- Climate & Sustainability ·
- Generative AI
-
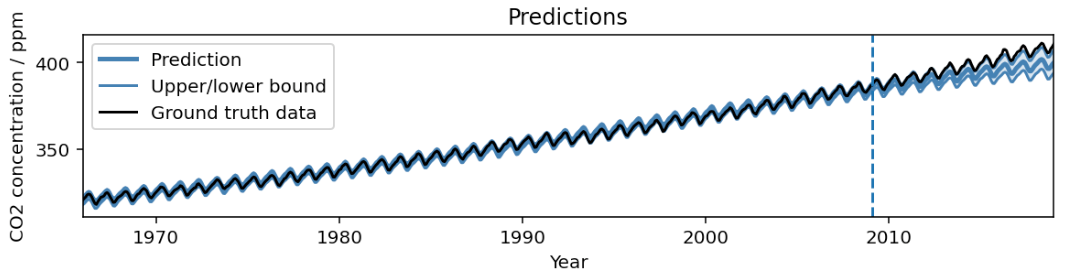
March 28, 2024
AutoBNN: Probabilistic time series forecasting with compositional bayesian neural networks- Algorithms & Theory ·
- Machine Intelligence ·
- Open Source Models & Datasets
-
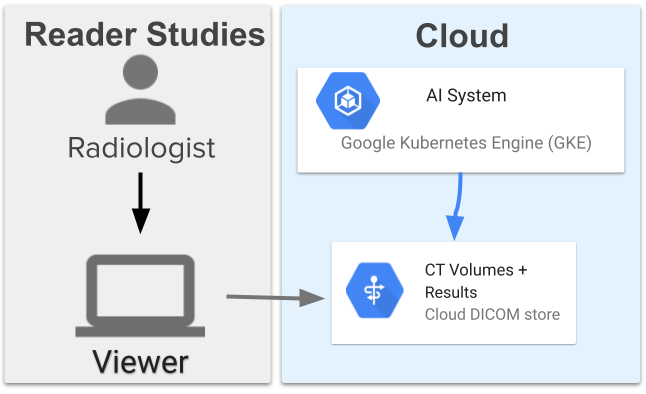
March 20, 2024
Computer-aided diagnosis for lung cancer screening- Health & Bioscience ·
- Human-Computer Interaction and Visualization ·
- Machine Intelligence
-

March 20, 2024
Using AI to expand global access to reliable flood forecasts- Climate & Sustainability ·
- Machine Intelligence